PEDIATRIC SPINAL ANOMALIES
Wallace W. Peck, MD, John R. Hesselink, MD,
& A. James Barkovich, MD
Most developmental disorders of the spine occur in either the upper cervical or lower thoracic and lumbar regions. These disorders result from defective embryogenesis of the spinal cord and vertebral column. They are found as isolated entities or in combination with one another, such as in Chiari malformation type II with an associated tethered cord and myelomeningocele. The age of clinical presentation ranges from birth to adulthood, depending on the type and severity of the malformation. MR imaging in the sagittal plane is particularly valuable for screening these patients, quickly yielding information about the position of the cerebellar tonsils and conus medullaris, the presence of syringohydromyelia, and any associated spinal dysraphic defects. Knowledge of normal development is very helpful for understanding congenital anomalies of the spine and spinal cord.
FORMATION OF THE SPINAL CORD
The spinal cord is formed from two parts. The segment extending from the medulla to the mid-lumbar enlargement develops by "neurulation." The more distal segment, including distal lumbar
cord, conus medullaris, and filum terminale, arise by "canalization and retrogressive differentiation."
![]()
Neurulation
On the fifth day of life a group of ectodermal cells proliferate to form the primitive streak along
the dorsal surface of the embryo. At the cephalic end of the streak is a rapidly proliferating group
of cells, surrounding a primitive pit, known as Hensens node. On day 15 and 16, cells enter the
primitive pit and migrate cephalad to form the notochordal process, which eventually becomes the
notochord. The notochord causes the overlying ectoderm to differentiate into the neural plate, which
is contiguous with the ectoderm. At 17 days the lateral portions of the neural plate thicken, forming
two neural folds with a neural groove between them. Contractile filaments concentrated in the
neuroepithelial cells of the neural folds begin to contract, bringing the edges together to form the
neural tube. Tube closure (Neurulation) begins at multiple sites and proceeds in both directions.
When neurulation is complete, the overlying ectoderm separates from the neural tube. The neural
crest cells migrate laterally to form the dorsal root ganglia and the sympathetic plexus. The anterior
neuropore (cephalic end) closes at about 24 days and the posterior neuropore (caudal end) closes at
about 27 days. The anterior neuropore closes at the lamina terminalis. The location of the posterior
neuropore is uncertain but probably resides in the lower lumbar region.
![]()
Canalization and Retrogressive Differentiation
Canalization is the process by which the neural tube elongates caudal to the posterior neuropore. During this process the notochord and neural epithelium fuse, forming a caudal cell mass. At 30 days multiple microcysts within this mass coalesce to form an ependyma-lined tubular structure that unites with the neural tube from above. This process is somewhat disorganized and may account for accessory lumina and ependymal rests found in the filum terminale and distal conus in adults.
At around 38 days of gestation, cell necrosis causes a decrease in the size of the caudal neural tube. This process of retrogressive differentiation leads to the formation of the distal conus medullaris, filum terminale, and the ventriculus terminalis (a focal dilatation of the distal central canal within the conus).1
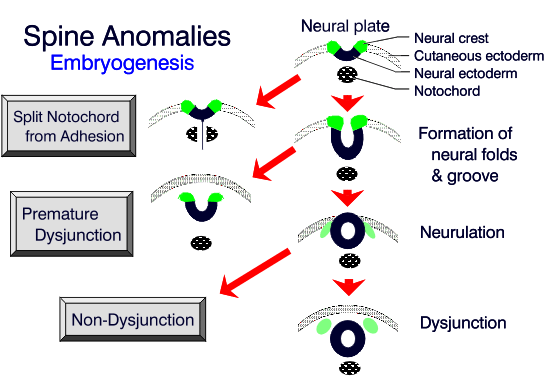
FORMATION OF THE VERTEBRAL COLUMN
Membrane Development
At around the 25th day the notochord separates from the gut and neural tube. Regional mesenchymal cells migrate into the areas ventral and dorsal to the notochord and form somites separated by intersegmental fissures. Each somite has a medial sclerotome and a lateral myotome portion. The medial sclerotome forms the vertebral body and the lateral myotome forms the paraspinal musculature. After separation of the neural tube from the superficial ectoderm, mesenchyme also migrates dorsally to form the posterior elements.
Chondrification
The sclerotomes then divide in half horizontally. The bottom half of one fuses with the top half of another to form the vertebrae. This occurs bilaterally forming both halves of the vertebrae. Notochordal remnants between the vertebrae become the nucleus pulposus within the intervertebral disk. Portions of the sclerotomes in the thoracic region migrate out to form the ribs.
Ossification
The chondral skeleton then ossifies. The formation of vertebrae at the caudal end is a little
disorganized (like neural tube formation). A mass of tissue composed of notochord, mesenchyme
and neural tissue divides into somites to form the sacral and coccygeal levels. This disorganized
development probably contributes to the frequent anomalous developments seen at this level.
![]()
ANOMALIES OF NOTOCHORD FORMATION
Diastematomyelia
In diastematomyelia the spinal cord is divided vertically into two hemicords, each with its own central canal and pia surrounding it. Females are affected more commonly (80%) than males. In two-thirds of cases the overlying skin shows nevi, hypertrichosis, lipomas, dimples or hemangiomas. Symptoms are nonspecific and similar to other causes of cord tethering.
This disorder may result from splitting of the notochord around an adhesion between the endoderm and the ectoderm. This split notochord might influence the formation of two neural tubes and
subsequently two hemicords. The notochord also influences the vertebrae formation and thus it is
common to have associated segmentation anomalies at the site of diastematomyelia.
![]()
The conus is low lying in 75% of cases. The hemicords frequently reunite below the cleft.
Hydromyelia is found in 50%. In 60% the two hemicords have a single dura surrounding them and
no spur or fibrous band. In the 40% that have two dural tubes, there is always a fibrous or bony spur
within the cleft. Bony spurs account for 60% while fibrous spurs account for 40%. The type of spur
is probably determined by the amount of trapped mesenchyme between the split notochord. On short
TR/TE images the osseous spur (unless it contains marrow) or a fibrous septum can be difficult to
distinguish from a CSF cleft that may be present between the two elements of separated spinal cord.
Gradient-echo or long TR/TE sequences that produce a myelographic effect often clarify the situation
because a dark spur or septum is contrasted against the bright CSF.
![]() ,
,
![]() In some cases, further
evaluation with plain CT may be necessary to accurately define the malformation.
In some cases, further
evaluation with plain CT may be necessary to accurately define the malformation.
Diastematomyelia accounts for 5% of congenital scoliosis and has been reported in 30-40% of myelomeningoceles. Kyphoscoliosis from osseous anomalies is seen in half of patients with diastematomyelia.
Split Notochord Syndrome
In this syndrome, persistent connection between the endoderm and ectoderm results in splitting or deviation of the notochord. In its most severe form, called a dorsal enteric fistula, a communication exists between the intestinal cavity and the dorsal skin in the midline. The fistulae traverse the prevertebral soft tissues, the vertebral bodies, the spinal canal and its contents, and the posterior elements. Any portion of this tract may involute or become fibrous, leaving fistulae or cysts. The dorsal enteric sinus, a remnant of the posterior portion of the tract, has an opening on the skin surface. Dorsal enteric cysts are trapped remnants of the middle portion of the tract found in the intraspinal or paraspinal compartments. The dorsal enteric diverticulum is a tubular diverticulum arising from bowel and represents a remnant of the anterior portion of the tract.1
Patients with dorsal enteric fistulae present as newborns with a bowel ostium exposed on their
back. Intraspinal enteric cysts usually present between 20 and 40 years as episodic local or radicular
pain that may progress to myelopathic symptoms. These are most commonly found in the lower
cervical or upper thoracic spine. The spinal canal is frequently enlarged. The cyst fluid may be
similar to CSF or xanthochromic/opaque. The cysts are usually ventrolateral. There is a male to
female predominance of 3:2.
![]()
ANOMALIES OF PREMATURE DYSJUNCTION
Spinal Lipomas
Fat and connective tissue masses are attached to the meninges or spinal cord. The fibrous component is greater near the junction of the cord and lipoma than at the junction of the lipoma and skin. Three types of spinal lipomas include the intradural lipomas (4%), lipomyelomeningoceles and lipomyeloceles (84%), and fibrolipomas of the filum terminale (12%).
Intradural lipomas and lipomyelomeningoceles are thought to be caused by premature separation of the neural ectoderm from the cutaneous ectoderm. This separation allows the mesenchyme to enter the ependyma-lined canal of the neural tube inducing fat formation. Fibrolipomas of the filum terminale probably result from an abnormality of retrogressive differentiation (see below).
Intradural lipomas account for less than 1% of intraspinal tumors. Only 25% present within the
first 5 years of life. They are juxta-medullary masses completely enclosed by dura. They occur most
commonly in the cervical and thoracic spine (66%), typically presenting with slow, ascending
monoparesis or paraparesis, spasticity, cutaneous sensory loss, and defective deep sensation. About
75% of lipomas are in the dorsal part of the cord. Only 2% have an associated hydromyelia and
syringomyelia. Lumbosacral lipomas typically present with flaccid paralysis of the legs and sphincter
dysfunction.
![]()

Lipomyelomeningoceles are lipomas attached to the dorsal surface of the neural placode. These
account for 20% of covered lumbosacral masses and slightly less than 50% of occult spinal dysraphism. Presentation is usually before 6 months of age and 50% are neurologically normal. There
may be a mass, sensory loss, bladder dysfunction, motor loss, foot deformities, and leg pain. A lipomyelocele consists of a lipoma attached to the cord and
subcutaneous tissue through a spina bifida but with a
normal sized subarachnoid space. Patients with
lipomyelomeningocele often have posterior herniation of
the malformed neural elements due to expansion of the
subarachnoid space ventral to the placode. Butterfly
vertebrae and segmentation anomalies are seen in 43%.
Sacral anomalies are found in 50% of patients and
include confluent foramina and partial sacral dysgenesis.
![]()
ANOMALIES OF NONDYSJUNCTION
These anomalies result from incomplete separation of neuroectoderm from cutaneous ectoderm. Anomalies of nondysjunction may be focal or diffuse. A focal anomaly would be a dorsal dermal sinus, while diffuse anomalies would include myelocele, myelomeningocele, and hemimyelocele.
Dorsal Dermal Sinuses
A form of occult spinal dysraphism, dorsal dermal sinuses are thin, epithelium-lined channels that
open on the skin posteriorly in a hyperpigmented patch or a hairy nevus. The sinus tracts extend deep
into the subcutaneous tissues, reaching the spinal canal in one-half to two-thirds of cases. The sinuses
may be attached to the dura, causing tenting of the thecal sac. When they pass intradurally, they may
end in the subarachnoid space, conus medullaris, filum terminale, a nerve root, a fibrous nodule on
the surface of the cord, or a dermoid or epidermoid cyst. Roughly 50% of the dermal sinuses end in
dermoid or epidermoid cysts. Conversely, 20% to 30% of dermoid cysts and dermoid tumors have
associated dermal sinus tracts. The course of the sinus tract may be short or long, and varies from
patient to patient. The tract may be lined by fat. The dermatome level of the sinus opening correlates
with the metameric level of the cord where it attaches.
![]()
Bony abnormalities vary from none when the tract penetrates at the level of a posterior ligament, to focal or multilevel spina bifida. When a dermoid or epidermoid is present, the nerve roots are frequently bound down to it. There may be a history of meningitis from extension of bacteria along the tract or from chemical irritation if the cyst ruptures.
T1 or T2-weighted sequences often demonstrate the sinus tract coursing obliquely through the subcutaneous tissues. Dermoids are identified by their fatty components. Although epidermoids can be isointense with CSF on all MR pulse sequences, usually they have a more heterogeneous internal texture than CSF. Flow-sensitive sequences can increase the contrast between free-flowing CSF and the solid epidermoid tumor. In equivocal cases, myelography with CT can help clarify the diagnosis.

Myelocele and Myelomeningocele
A type of spina bifida aperta, myelocele and myelomeningocele result from localized failure of closure of the neural tube. The neural folds remain in continuity with the cutaneous ectoderm at the skin surface, forming the neural placode. The mesenchyme destined to form the posterior elements remains trapped laterally, causing a wide spina bifida.
Anomalies of vertebral segmentation or hemivertebrae are commonly present, resulting in short radius kyphoscoliosis in approximately one-third. Another 65% develop kyphoscoliosis as a result of neuromuscular imbalance.
The spinal cord is always tethered. Aside from fetal
ultrasound, imaging studies usually are not performed because
early surgical closure of the open spinal cord is most critical to
avoid further damage to the neural elements. The patients typically have a stable neurologic defect
unless other associated anomalies cause problems. If preoperative imaging is done, a number of
features require definition, such as the location of the neural placode, fibrovascular tethering band(s),
ventral and dorsal roots, dorsal root entry zones, and any nerve roots crossing in an aberrant fashion.
It may be difficult to determine whether a low-lying placode in a dorsal meningocele is actually
tethered or simply positioned in the meningocele as a result of more cephalad tethering by the
fibrovascular band.
![]() Possible associated anomalies include syringohydromyelia (found in 40% of
Arnold Chiari II patients), diastematomyelia (30%-45% of myelomeningoceles) lipoma, arachnoid
cyst, dermoid, or epidermoid.
Possible associated anomalies include syringohydromyelia (found in 40% of
Arnold Chiari II patients), diastematomyelia (30%-45% of myelomeningoceles) lipoma, arachnoid
cyst, dermoid, or epidermoid.
Imaging of the postoperative myelomeningocele spine is usually performed because of deterioration of neurological function. When imaging these patients, one must look for postoperative hematoma or complicating infection, compression of cord by residual or recurrent tumor, cord ischemia or infarction, myelomalacia, arachnoid cyst, diastematomyelia, and re-expansion of a syringohydromyelia. T2-weighted coronal images are helpful to rule out occult diastematomyelia. Focal cord narrowing may occur if the dura was pulled too tight at surgery. Symptomatic re-tethering is a diagnosis of exclusion. Although retethering is a clinical diagnosis, MR signs of retethering by scar include angulation or kinking of the cord, a straightened or taut-appearing cord, or a direct cord interface with dural or epidural structures. Because of the spectrum of abnormalities associated with myelomeningoceles, imaging of at least the lumbar and thoracic (and possibly the entire) spine should be performed in the postoperative patient. If the spine is normal, imaging of the brain should be considered to rule out hydrocephalus or shunt malfunction.
Hemimyelocele
Between 30% and 45% of patients with myelomeningoceles will have associated diastematomyelia. The cord may be split above, below, or at the same level as the myelomeningocele. When one of the hemicords has a small myelomeningocele it is known as hemimyelocele. This occurs in 10% of all myelomeningoceles. The defect is usually off the midline. If the hemicords are asymmetric, the smaller hemicord is usually ventral. The child typically only has impaired function on the side of the hemimyelocele.
Chiari II Malformation
An anomaly of the cervical spinal cord, brain stem, and hind brain, the Chiari II malformation is observed in varying degrees in all patient with myelomeningoceles. Occasionally a Chiari II malformation may be seen without a myelomeningocele.
There are many theories of the genesis of this malformation. It is generally accepted that the
posterior fossa is too small, causing herniation of the brainstem and cerebellar tonsils into the upper
cervical spinal canal. One unifying theory suggests that the posterior neuropore remains open too
long, thereby decompressing the ventricular system and allowing the bony posterior fossa to close
too early forming a small compartment. This alteration in closure of the posterior neuropore is the
myelomeningocele. Upon surgical closure the infants typically develop hydrocephalus within 48
hours.
![]() Many anomalies of the skull, brain, and spine can be found with Chiari II malformations.
Many anomalies of the skull, brain, and spine can be found with Chiari II malformations.
ANOMALIES OF THE CAUDAL CELL MASS
These anomalies result from aberrant canalization and retrogressive differentiation. At 8 weeks
of gestation the conus medullaris extends all the way to the end of the spinal canal. As the embryo
matures the distal portion undergoes retrogressive differentiation and the vertebral column lengthens
more rapidly than the remaining cord. As a result, by 24 weeks of gestation the conus is at the
bottom of S1, at birth it is probably at L3, and by 3 months of age the conus is usually at L1-2 where
it stays through adulthood. In a large series of patients the conus was found above the L2-3 level in
97.8% of cases.
![]() Thus, if the conus lies at or below L3, a careful search should be made for a source
of tethering.
Thus, if the conus lies at or below L3, a careful search should be made for a source
of tethering.
Tight Filum Terminale Syndrome
Incomplete retrogressive differentiation is most likely responsible for this anomaly. A complex
of neurologic and orthopedic deformities is associated with a short, thick filum terminale and a low
position of the conus medullaris. Muscle stiffness or weakness and abnormal lower extremity reflexes
are common. There may be bladder dysfunction, sensory changes, orthopedic deformities (i.e. club
foot), back pain, and radiculopathy. Scoliosis is frequently present. Age of onset of symptoms is
variable.
![]()
The normal filum is ≤ 2mm in diameter at L5-S1. About 55% of patients with tethered cord
syndrome have an obviously thick, fibrotic filum and 23% have a small fibrolipoma within the
thickened filum. Usually the conus is low lying (below L2-3), but occasionally it is at a normal
position. In 25% one may find a small area of CSF signal within the conus. This may represent a
small hydromyelia or a normal physiologic finding from the tethering. The fluid signal typically
disappears after untethering.
![]()
Sagittal T1 or T2-weighted images readily show the level of the conus. A thickened filum may be difficult to distinguish on MR, especially when the conus terminates at a normal level. Thickening of the filum can be simulated by normally clumped ventral and dorsal roots situated in the posterior aspect of the thecal sac. Serial axial views are best for displaying the anatomy of the distal conus and cauda equina. Any associated fibrolipoma is easily seen on T1-weighted scans.
Syndrome of Caudal Regression
This anomaly is probably caused by a disturbance of the caudal mesoderm before the fourth week of gestation. Faulty retrogressive differentiation results in an abnormal distal spinal cord, and associated maldevelopment of the notochord results in various vertebral anomalies. The incidence is 1 in 7,500 live births. One in six (17%) have diabetic mothers.
The degree of maldevelopment is often profound and the clinical deficits severe. The spectrum of anomalies seen includes: fusion of the lower extremities (sirenomelia), lumbosacral agenesis, anal atresia, malformed external genitalia, bilateral renal aplasia, and pulmonary hypoplasia with Potters facies. Motor deficits in the lower extremities are more severe than sensory changes. Almost all have a neurogenic bladder. Children with mild signs and symptoms usually are evaluated for possible tethered cord.1
The degree of spinal agenesis is variable, ranging from partial sacral agenesis to complete agenesis
of the lumbar and sacral spine. The spinal canal is frequently severely stenotic just above the last
intact vertebrae. The cord has a typical blunt or wedge-shaped appearance. There can be a tethering
lipoma or lipomyelomeningocele.
![]() MR imaging in the sagittal plane effectively shows the missing
components of the lumbosacral spine.
MR imaging in the sagittal plane effectively shows the missing
components of the lumbosacral spine.
Fibrolipomas of the Filum Terminale
Isolated fibrolipoma is a minor anomaly in development of the filum terminale. The filum is a
long, fibrous band that extends from the conus down through the subarachnoid space and dura to
insert on the dorsal aspect of the first coccygeal segment. These lesions are often asymptomatic. One
study incidentally found them in 6% of autopsy studies. They may be found with the tight filum
terminale syndrome described above. Some patients may not present until late into adult life. Once
detected, these patients should be followed for signs of tethering.
![]()
Anterior Sacral Meningoceles
Focal erosion or dysgenesis of segments of the sacrum and coccyx permit herniation of a CSF-filled meningeal sac anteriorly into the pelvis. Nerve roots may enter the sac. Patients usually present in the second or third decades of life. Symptoms result from pressure on adjacent structures (i.e., constipation, urinary frequency and incontinence, dyspareunia, numbness or paresthesias in the sacral dermatomes). Headaches may occur from fluid shifts within the spinal subarachnoid space. The embryogenesis is unknown.
Sacrococcygeal Teratomas
These rare congenital tumors probably arise from primitive, multipotential cells along the primitive streak. Two thirds are of the mature variety and one third are of the immature or anaplastic type. They are usually well-encapsulated, lobulated tumors with both solid and cystic components. These teratomas have been classified by their location:
Type I (47%): almost entirely posteriorly.
Type II (35%): large posterior mass with significant pelvic extension.
Type II (8%): small posterior mass but mainly within pelvis and abdomen.
Type IV (10%): entirely presacral.
Direct involvement of the sacral canal is rare. Patients present with a mass and/or urinary/bowel
problems. The MR appearance is variable depending upon its composition. Typically the tumor is
heterogeneous from the high signal fat, the intermediate signal soft tissue, and the low signal from
calcium.
![]()
ANOMALIES OF UNKNOWN ORIGIN
Myelocystocele
This condition is probably a rare variant of a myelomeningocele. It includes an occult spinal
dysraphism in which a hydromyelic spinal cord and arachnoid are herniated through a posterior spina
bifida. These reportedly occur most commonly in the lumbosacral region and may be associated with
anorectal and urogenital malformations (e.g., cloacal exstrophy). They can rarely occur at higher
levels.
![]()
Simple Meningoceles
Posterior herniation of dura, arachnoid, and CSF produces a cyst-like mass in the subcutaneous tissues of the back. Localized bony defect is typically present. Imaging is obtained to determine the size, shape, presence of nervous tissue, and its relationship to the conus.
Lateral Meningoceles
In this case CSF-filled protrusions of dura and arachnoid extend out through enlarged neural foramina. About 85% of thoracic meningoceles are associated with neurofibromatosis. There may be scoliosis present, convex toward the lateral meningocele. They may disappear after shunting dilated ventricles.1
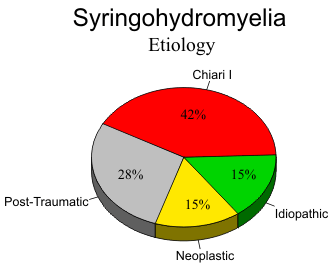
Syringohydromyelia
Syringomyelia refers to a cavity in the spinal cord extending lateral to or independent of the central canal. Hydromyelia refers to a dilated central canal of the cord. Most cavities involve both parenchyma and central canal, and the term syringohydromyelia reflects this combined phenomenon. Many use syrinx and syringomyelia as general terms for any cord cyst.
The clinical presentation is variable depending on the location and extent of the syrinx. The classic picture is segmental weakness and atrophy of the hands and arms, with loss of reflexes and segmental anesthesia. However, typical symptoms are unilateral, confined to the lower extremities, or absent. Accompanying severe pain can be debilitating. The symptoms are unpredictable with periods of waxing and waning and may mimic brainstem compression.
There are several theories regarding the formation of syringohydromyelia. Gardner's theory
suggests that there is a lack of perforation of the foramen of Magendie that forces CSF through the
obex into the central canal of the cord.
![]() Williams proposes that there is free flow of CSF upward
from the spinal subarachnoid space into the intracranial cisterns, but flow is partially blocked in the
reverse direction.
Williams proposes that there is free flow of CSF upward
from the spinal subarachnoid space into the intracranial cisterns, but flow is partially blocked in the
reverse direction.
![]() He suggests that this cranial-spinal pressure difference leads to CSF being sucked
into the central canal of the cord from the 4th ventricle, creating a syrinx. Williams also proposes that
the fluid within the syrinx moves both up and down as a result of changes in epidural venous pressure.
This may result in extension of the syrinx cavity. Ball and Dayan,
He suggests that this cranial-spinal pressure difference leads to CSF being sucked
into the central canal of the cord from the 4th ventricle, creating a syrinx. Williams also proposes that
the fluid within the syrinx moves both up and down as a result of changes in epidural venous pressure.
This may result in extension of the syrinx cavity. Ball and Dayan,
![]() as well as Aboulker,
as well as Aboulker,
![]() believe that
the craniospinal pressure gradient is present but reversed in direction from that proposed by Williams.
These authors believe that increased CSF pressure in the spinal CSF space results in CSF filtering
from the subarachnoid space into the central canal through the spinal cord. Since passage into the
IV ventricle is blocked, a syrinx forms.
believe that
the craniospinal pressure gradient is present but reversed in direction from that proposed by Williams.
These authors believe that increased CSF pressure in the spinal CSF space results in CSF filtering
from the subarachnoid space into the central canal through the spinal cord. Since passage into the
IV ventricle is blocked, a syrinx forms.

Direct evaluation of the cord and the entire spinal neuraxis with MR provides the most sensitive
and specific evaluation of syringohydromyelia. In the largest series by Sherman and his group,
![]() four
separate groups of patients were identified based on the appearance of the cavities and any associated
anomaly or relevant history. Approximately 41 per cent had syringomyelia associated with tonsillar
ectopia (Chiari I malformation), 28 per cent had posttraumatic syrinx, 15 per cent were associated
with neoplasm and 15 per cent were idiopathic. When the cavities were analyzed for specific
characteristics, a few observations were made. At least one third of patients with nonneoplastic
cavities had associated high-signal intensity in the cord contiguous to the cavity or in the parenchyma
distal or proximal to the cavity. This high-signal intensity in a simple syrinx is speculated to represent
gliosis, edema, demyelination or microcyst malformation.
four
separate groups of patients were identified based on the appearance of the cavities and any associated
anomaly or relevant history. Approximately 41 per cent had syringomyelia associated with tonsillar
ectopia (Chiari I malformation), 28 per cent had posttraumatic syrinx, 15 per cent were associated
with neoplasm and 15 per cent were idiopathic. When the cavities were analyzed for specific
characteristics, a few observations were made. At least one third of patients with nonneoplastic
cavities had associated high-signal intensity in the cord contiguous to the cavity or in the parenchyma
distal or proximal to the cavity. This high-signal intensity in a simple syrinx is speculated to represent
gliosis, edema, demyelination or microcyst malformation.
![]() The presence of edema is supported by
the return of the hyperintense parenchyma to
normal signal intensity following myelotomy and
shunt decompression of the cyst.
The presence of edema is supported by
the return of the hyperintense parenchyma to
normal signal intensity following myelotomy and
shunt decompression of the cyst.
![]()
Enhancement with gadolinium increases the certainty of distinguishing a primary syrinx from a cystic neoplasm. If a cavity is noted within the cord on screening MR sequences, gadolinium should be given and T1-weighted scans acquired in sagittal and axial planes. If no enhancing nodule is seen, a cord neoplasm is unlikely.
Examination of the entire spinal cord with sagittal images is mandatory. In regions where a cavity is suspected but somewhat equivocal in the sagittal projection, axial scans should be done. Failure to examine the entire spinal neuraxis may result in missing cavities that are separated from the primary or larger cavity. Long segments of normal appearing cord may intervene between a cervical cavity and one located in the conus medullaris. Failure to recognize an additional cavity could result in inappropriate or incomplete shunting and subsequent failure of the patient to improve clinically.
REFERENCES